por Jessika Viana
O desenvolvimento de medicamentos pode ser comparado a um triatlo, que apresenta diversas etapas a serem cumpridas. É de suma importância que um composto protótipo passe por todas elas para que se torne um medicamento. Porém, esse era um processo que demorava várias décadas. E, como dito, era! Nos últimos anos os cientistas têm utilizado técnicas computacionais cada vez mais poderosas para processar a grande quantidade de dados biológicos e químicos disponíveis, além de algoritmos cada vez mais robustos na busca de novos agentes terapêuticos.
Pense no seguinte: no desenvolvimento de medicamentos um composto químico deve primeiro passar por uma série de etapas que focam no estudo de uma determinada doença como, por exemplo, o câncer. Em seguida, os cientistas identificam os pontos chaves desta doença, ou seja, identificam os mecanismos celulares e moleculares envolvidos na sua ocorrência. Vamos supor, por exemplo, que um tipo específico de câncer seja caracterizado pela presença de uma proteína mutada. Após essa identificação, pode-se avaliar in vitro a atuação de um composto químico frente a esta proteína mutada, em uma tentativa de inibir o efeito danoso da proteína e reverter sua ação no organismo, podendo levar a uma regressão do câncer.
Em seguida, sendo confirmado o efeito do composto químico, que passa a ser chamado de composto protótipo ativo, ele passará por testes em organismos vivos (como pequenos roedores) e por estudos pré-clínicos e clínicos em seres humanos a fim de avaliar sua eficácia. Se a ação inibitória do composto se mantiver e apresentar baixa ou quase nenhuma toxicidade ao ser humano, este pode se tornar um medicamento.
Este composto inibidor, antes da utilização das técnicas computacionais, deveria ser primeiro sintetizado em laboratório para então passar pelas etapas de testes, demandando um grande gasto de tempo e de dinheiro para construção de um esqueleto molecular que não se sabia se seria ativo ou não Ainda, para um total de 10.000 compostos estudados, apenas um se torna medicamento, pois a maior parte dos compostos é eliminada por não apresentarem atividade biológica ideal ou apresentarem efeitos colaterais inaceitáveis Assim, antes que um novo medicamento receba aprovação regulatória, ele precisa passar por testes altamente complexos, um processo demorado, que pode levar de 10 a 17 anos! E que, no entanto, pode ser acelerado com o uso de soluções computacionais.
Com o advento das técnicas computacionais é possível realizar diversas simulações para se identificar compostos candidatos, possivelmente bioativos, para uma doença alvo. Esses avanços têm evoluído em conjunto com o conhecimento da biologia molecular, equipamentos de última geração e técnicas da computação e automação.
A pesquisa farmacêutica pela descoberta de medicamentos inicia com um grande foco na estrutura molecular de um alvo biológico de uma doença, que pode ser uma membrana, uma proteína, um DNA ou RNA, sendo estes conhecidos como macromoléculas biológicas. Por meio do sequenciamento do genoma, a luta contra diversas doenças vêm sendo conquistadas, já que ele auxilia na identificação de macromoléculas biológicas essenciais no desencadeamento de uma doença. Este sequenciamento expandiu a era big data, favorecendo o surgimento de diversos bancos de dados aplicados ao desenvolvimento de fármacos, como o banco de dados de proteínas, banco de dados de pequenas moléculas e banco de dados de genomas.
Uma vez que um alvo biológico com papel essencial no desenvolvimento da doença é identificado, os estudos computacionais podem se concentrar neste alvo para encontrar um medicamento que reduza, ou mesmo, cure uma doença. A esta área de desenvolvimento computacional de um novo medicamento denomina-se desenho de medicamentos auxiliado por computador (do inglês Computer Aided Drug Design - CADD).
O CADD tem como objetivo principal prever se um determinado composto químico apresenta características químicas favoráveis ou não, além de avaliar como e com que intensidade um composto pode interagir com um alvo biológico.
Para realizar esses estudos, uma das abordagens consiste dos pesquisadores acessarem bancos de dados contendo milhões de estruturas moleculares virtuais que podem ser facilmente sintetizadas e avaliarem, através de simulações computacionais, a sua interação em partes específicas de uma proteína, conhecido como sítio ativo. Através desta simulação é possível identificar bons candidatos a fármaco selecionando aqueles que apresentaram melhor acoplamento com a proteína. Este método é comumente conhecido como atracamento ou ancoramento molecular (do inglês molecular docking), método este onde os novos compostos bioativos são posicionados dentro do alvo biológico.
Uma segunda técnica identifica a atividade biológica de um composto a partir de uma comparação de características químicas com medicamentos já existentes. Nesta abordagem também é possível identificar qual característica físico-química do composto é mais importante para uma boa atividade biológica. A esta segunda técnica denomina-se relação quantitativa estrutura-atividade biológica (do inglês Quantitative Structure-Activity Relationships - QSAR), método este onde os novos compostos são baseados na estrutura de compostos bioativos pré-existentes.
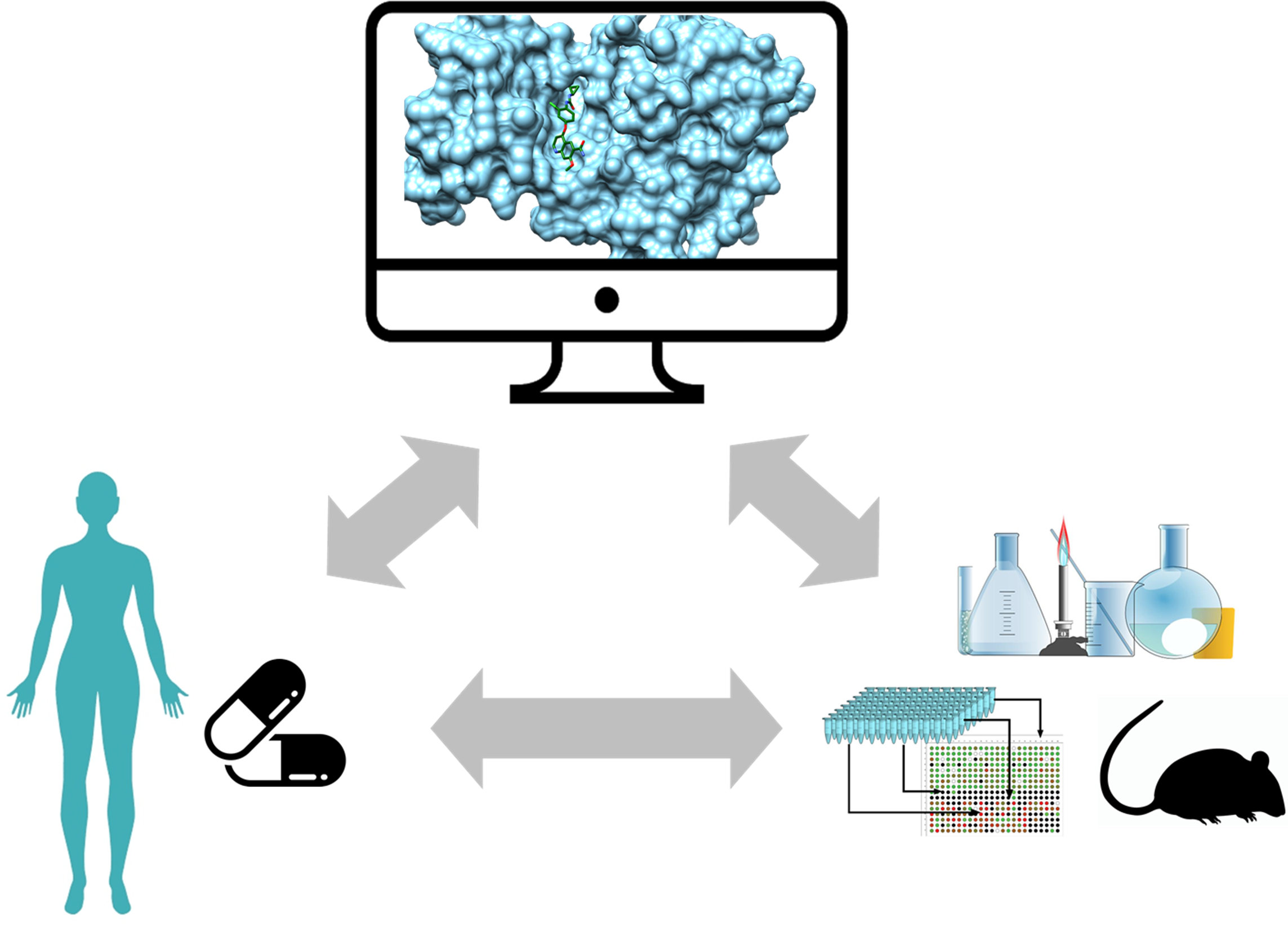
<Legenda: Representação esquemática das metodologias aplicáveis a bioinformática e desenvolvimento de fármacos.>
Podemos citar como exemplos de medicamentos descobertos através da utilização do CADD o Captopril (anti-hipertensivo), Dorzolamide (controle da pressão intraocular), Saquinavir (antiviral), Zanamivir (antiviral), Oseltamivir (antiviral), Aliskiren (anti-hipertensivo). Outro exemplo é o Rogaratinibe, que atualmente está em testes clínicos de fase 2 e 3 com ação inibitória da proteína FGFR (do inglês Fibroblast Growth Factor Receptors). A FGFR é uma proteína mutada que está presente em pacientes com carcinoma urotelial metastático, um tipo específico de câncer urotelial. Nestes estudos, compostos com estrutura similar ao de Rogaratinibe são avaliados para identificar quais outras características químicas podem fornecer uma maior eficácia frente ao mesmo tipo de câncer.
Além destas técnicas, a Bioinformática tem auxiliado na busca por sítio ativo de proteínas, geração de estruturas tridimensionais de pequenos ligantes (fármacos, cofatores) e proteína. Uma terceira abordagem apresenta ferramentas poderosas de simulação computacional de movimento de átomos, sendo possível observar como se comportaria uma proteína quando exposto a um fármaco em um meio aquoso.
Desta forma, os computadores adquirem um papel auxiliar na decisão de se ter ou não um composto químico promissor, o que por sua vez ajuda na redução do custo e do tempo no desenvolvimento de um fármaco. Contudo, os cientistas se mantêm atentos e cautelosos quanto aos resultados produzidos por estes métodos computacionais, uma vez que as simulações ainda representam pouco a realidade biológica, apresentando algumas lacunas em cálculos teóricos que ainda estão em desenvolvimento. Portanto, é fundamental a progressão neste campo de pesquisa. Com a recente implementação de novos métodos de predição da estrutura tridimensional de proteína baseado na aprendizagem profunda, certamente haveremos de observar em breve novos avanços nesta área.
Referências:
- GORE, M.; JAGTAP, U. B. (Ed.). Computational drug discovery and design. Humana Press, 2018.
- COLLIN, M. P.; LOBELL, M.; HÜBSCH, W. et al. Discovery of Rogaratinib (BAY 1163877): a pan‐FGFR Inhibitor. Chem Med Chem, v. 13, n. 5, p. 437-445, 2018.
- KAIRYS, V.; BARANAUSKIENE, L.; KAZLAUSKIENE, M. et al. Binding affinity in drug design: experimental and computational techniques. Expert opinion on drug discovery, v. 14, n. 8, p. 755-768, 2019.